Hands-On: Loading Our Input Dataset Onto Galaxy
Overview
Teaching: min
Exercises: 60 minQuestions
Objectives
Locate a sequencing dataset from NCBI based on info from a publication
Download sequencing metadata in bulk from NCBI
Load data from the NCBI SRA into the environment
Prerequisites
This tutorial assumes that you have done the following:
- Signed up for a Galaxy account and can log in.
- Have taken a look at the Galaxy Environment 101 Tutorial and are familiar with the concepts therein.
- Have familiarized yourself with the steps of variant-calling and related file-formats (you can always do this as you progress through the tutorial as needed!).
Obtaining data from the NCBI Short Read Archive
First we need to identify some samples of interest. The Sequence Read Archive (SRA) is the primary archive of unassembled reads operated by the US National Institutes of Health (NIH). SRA is a great place to get the sequencing data that underlie publications and studies. We know where to find the data because Science (and in fact, most publications) require authors to deposit their data in a public database and give us the information we need to find it on the database.
The data availability statement can be found in the acknowledgements section of Lemieux et al. 2020:

As you can see, we now know we can find sequencing data related to this study under BioProject PRJNA622837. As defined by the NCBI, a BioProject is “a collection of biological data related to a single initiative, originating from a single organization or from a consortium”, so they generally contain multiple sequencing data sets. The NCBI has much a much more in-depth description of BioProjects on its webpage, but their general structure can be summarized below:
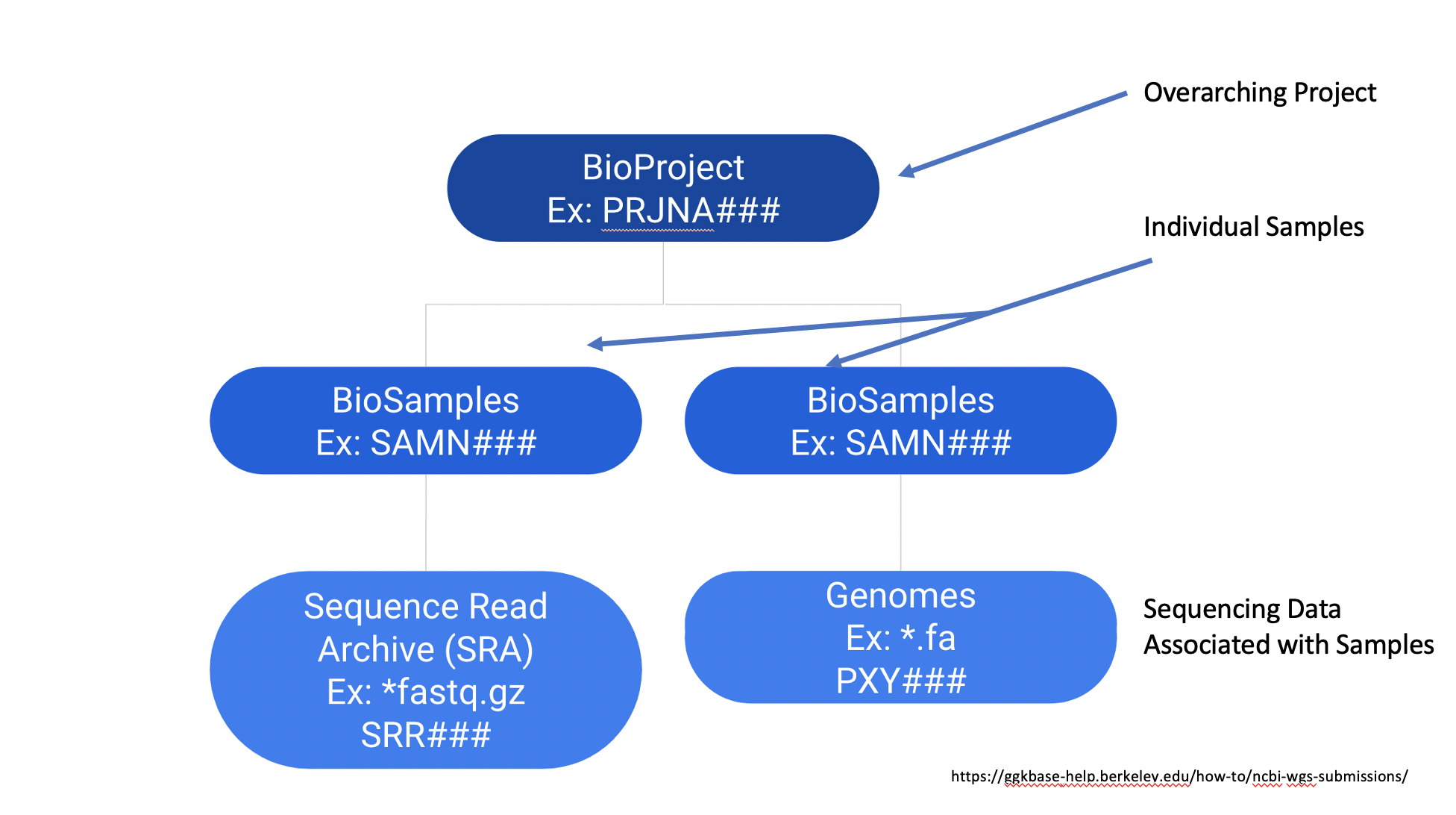
“Hands-On: Get Metadata from NCBI SRA”
- Go to NCBI’s SRA page by pointing your browser to
https://www.ncbi.nlm.nih.gov/sra- Perform a search using the Bioproject ID from the paper:
BioProject PRJNA622837- The web page will show a large number of available SRA datasets related to project described in Lemieux et al. 2020 (at the time of writing there were 22,173).
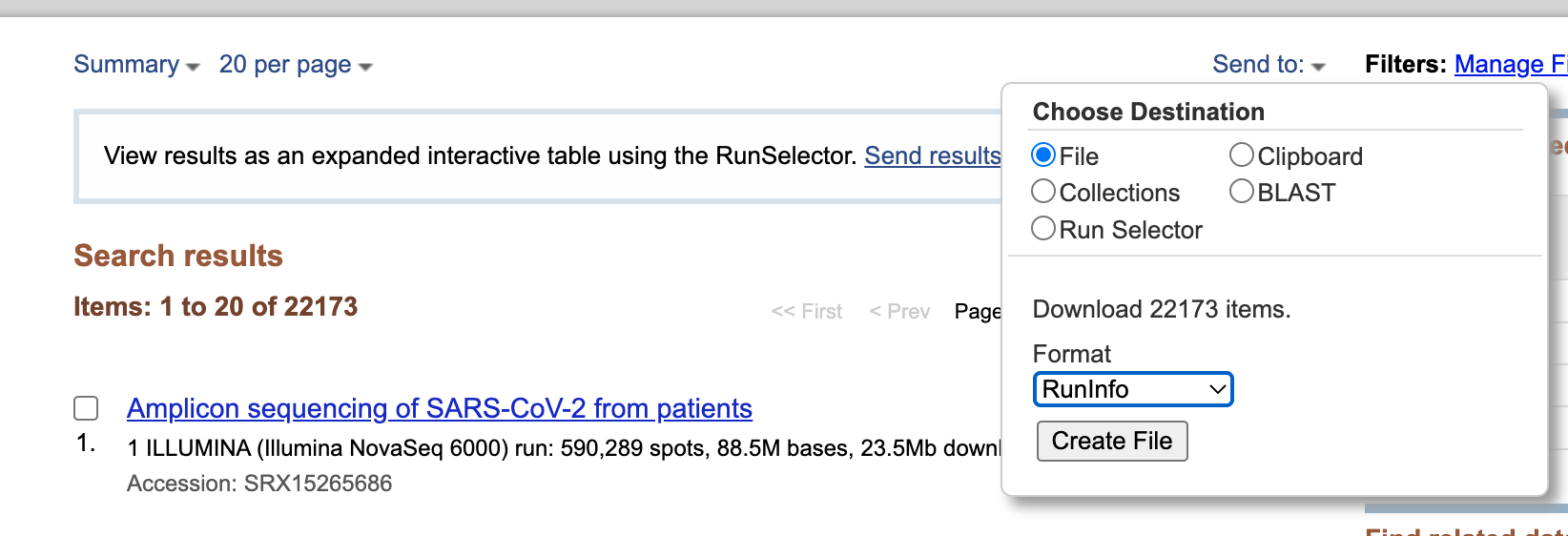
- Download metadata describing these datasets by:
- clicking on Send to: dropdown
- Selecting
File- Changing Format to
RunInfo- Clicking Create file. The screen should now look like this:
- Click “Create File” to download the
SraRunInfo.csvfile to a place on your computer where you will be able to find it easily for the next step.

Getting Information About Individual Samples
So, what is in the the SRA Run Info file?
The file we just downloaded is not sequencing data itself. Rather, it is metadata describing the properties of sequencing reads. We could actually open this file in a program like Excel, and if we did so, we could see columns of information like Release Date, Sequencing Platform, and Sample Name as reported by the researchers. In this study every accession corresponds to an individual patient whose samples were sequenced. We will filter this list down to just a few sequenced samples that will be used in the remainder of this tutorial.
This is all very valuable information - for example, if we want to know more about the sample that became the SRA sequencing dataset SRS8612691 we can use the Sample Name column to retrieve information about when the sample was actually collected. You can search for a Sample Name in the general NCBI database by using the All Databases option in the toolbar:

We can see that the sample that was sequenced to become the SRA sequencing dataset SRS8612691 was collected on January 4th, 2021 and was collected as part of routine surveillance:

Processing and Filtering Metadata on Galaxy
Hands-On: Upload
SRARunInfo.csvonto Galaxy
- If you haven’t already, log in to your Galaxy account. Create a new, empty history called something like “Variant Calling Tutorial”.
- In the tools panel, click the Upload Data button:
- Find
SRARunInfo.csvon your computer, and drag it into the “Drop Files Here” space in the dialog box that pops up.- Click the Start button to begin the upload.
- At this point, you can press the Close button. In a several seconds,
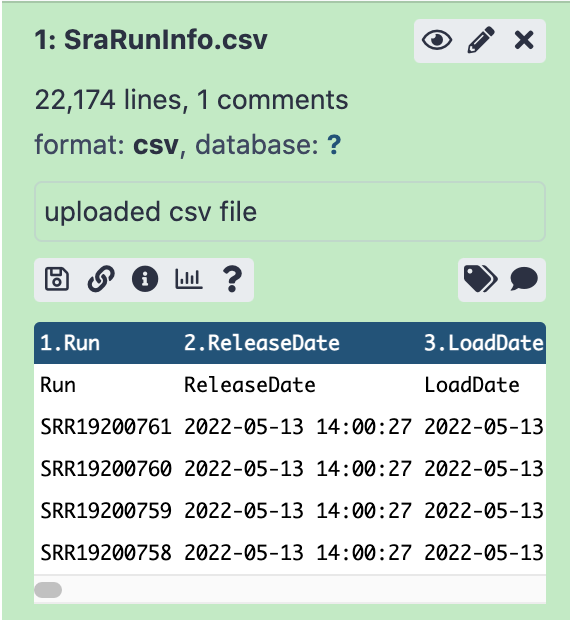
SRARunInfo.csvshould be ready to look at in the History panel.- You can now look at the content of this file by clicking on the eye icon . You will see that this file contains a lot of information about individual SRA accessions.
How large is the sequencing file associated with the accession number
SRR14114810?Solution
272 MB. If you open the file by clicking the eye icon, you should be able to find
SRR14114810in the Run column, and then look over to see 272 in the size_MB column.Note: If you ever want a smaller summary of the data, you can also click the name of of item in your history, which should produce something like this:
The Galaxy servers MIGHT powerful enough to process all 22,000+ datasets, but to make this tutorial bearable we need to selected a smaller, but still interesting, subset. In particular, we are interested in samples from early in the ongoing Covid-19 pandemic, so we will be choosing samples collected in April and May of 2020, which I have chosen using the “Collection Date” information retrieved by as described above.
We are going to focus on the following two sequencing data sets:
- Run Number
SRR12733957: A sequencing run from a sample collected on April 6th, 2020. - Run Number
SRR11954102: A sequencing run from a sample collected on May 2nd, 2020.
Warning: Don’t get caught by the wrong “Cut”!
WARNING: There are two cut tools in Galaxy due to historical reasons. For this tutorial we are assuming you are using the named EXACTLY The other tool follows a similar logic but with a different interface.
Hands-On: Creating a subset of data
Find the tool in the Filter and Sort section of the tool panel. You may find that Galaxy has an overwhelming amount of tools installed. To find a specific tool type the tool name in the tool panel search box to find the tool.
Make sure the SraRunInfo.csv dataset we just uploaded is listed in the Select lines from field of the tool form.
- In the Pattern field enter the following expression:
SRR12733957|SRR11954102. The “|” symbol (called a “pipe”) means “or”. So we are telling this tool to find lines containingSRR12733957ORSRR11954102.- Click the
Executebutton.- Once the this has been executed, you should have a file with a total of three lines. If you look at the file (), you will see that one of the two lines has been duplicated to take the place of the “header” line (which is fine for now).
- Cut the first column from the file using the tool, which you will find in Text Manipulation section of the tool pane.
- Make sure the dataset produced by the previous step is selected in the File to cut field of the tool form.
- Change Delimited by to
Comma.- In List of fields drop-down menu select
Column: 1. This is telling the tool to only return the first column of our data.- Hit
Execute. This will produce a text file with just two lines:SRR12733957 SRR11954102
Downloading the actual sequence data
So, now we have a file that contains just the two accession numbers for the sequencing data sets that we want. This is the perfect input for tools in Galaxy that can use this information to download big sequencing data sets straight from the NCBI SRA directly into your Galaxy environment without ever putting the big files on your computer.
Hands-On: Getting data from SRA
- with the following parameters:
- Select Input Type:
List of SRA Accession, one per line.- The input parameter select input type should point the output of .
- Click the
Executebutton. This will run the tool, which retrieves the sequence read read datasets and places them into your Galaxy environment. Note that this step can take a few minutes, so this might be a good time get get a fresh cup of coffee!- Take a look at the entries that were created in your history panel:
Pair-end data (fasterq-dump): Contains Paired-end datasets (if available)Single-end data (fasterq-dump): Contains Single-end datasets (if available)Other data (fasterq-dump): Contains Unpaired datasets (if available)fasterq-dump log: Contains Information about the tool executionIs the data we downloaded single-end or paired-end data?
Solution
Our data is paired-end! If you click each of the data files generated that are now in your history, you will see that
Single-end data (fasterq-dump)is an empty list, meaning that the tool did not download any files of that type. On the other hand,Pair-end data (fasterq-dump)is a “list of pairs with 2 items”, each of which is a FASTQ-formatted file if you look inside:
Pair-end data (fasterq-dump), Single-end data (fasterq-dump) and Other data (fasterq-dump) are actually collections of datasets. Collections in Galaxy are logical groupings of datasets that reflect the semantic relationships between them in the experiment / analysis. In this case the tool creates separate collections for paired-end reads, single reads, and other (any other type of file). See the Galaxy Collections tutorial and watch Galaxy tutorial videos (with names beginning with “Dataset Collections”) for more information.
Explore the collections by first clicking on the collection name in the history panel. This takes you inside the collection and shows you the datasets in it. You can then navigate back to the outer level of your history.
Once fasterq finishes transferring data (all boxes are green / done), we are ready to analyze it.
SRA Upload Workaround: Getting data from a Galaxy History
When to use this tool: If you have waited for more than 12 hours for these FASTQ files to successfully load into your Galaxy history using the tool , you can transfer these files from an existing history I set up for this week.
- Navigate to
https://usegalaxy.org/u/faes.biof521/h/biof521sp22week2tutorialfiles(the URL will depend on the exact history you are trying to import).- In the top right-hand corner, click the big ”+” button for the option to import this history into your Galaxy account.
- Once that is done (it should just take a few moments), Galaxy will automatically take you back to your home screen, with the imported history now active.
- Now you can go to the
View All Historiesoption, and you should see this history side by side with your older histories.- Drag and drop only the files that you need (in this case, probably just the
paired-endcollection into the history you set up for this tutorial.- Once you are done with that, make sure to click the Switch To tab above your Week 2 Tutorial history to make that one active before pressing the Home icon on the top bar to return to your usual Galaxy view.

What does our paired-end data actually look like?
It is common to prepare pair-end and mate-pair sequencing libraries. This is highly beneficial for a number of applications discussed, such as those discussed in Week 2’s Genome Assembly module. For now let’s just briefly discuss what these are and how they manifest themselves in FASTQ form.
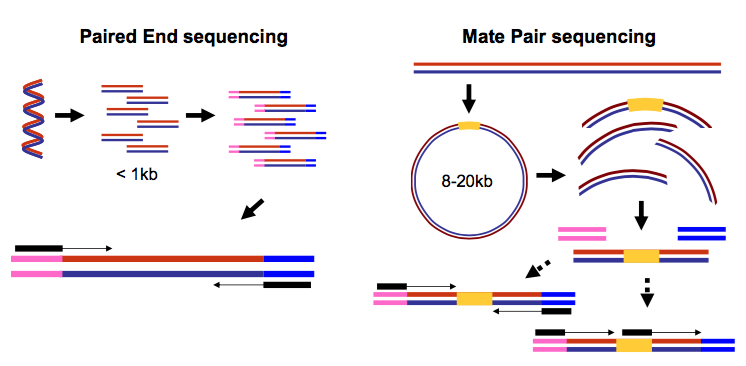
In paired-end sequencing (left) the actual ends of rather short DNA molecules (less than 1kb) are determined, while for mate-pair sequencing (right) the ends of long molecules are joined and prepared in special sequencing libraries. In these mate pair protocols, the ends of long, size-selected molecules are connected with an internal adapter sequence (i.e. linker, yellow) in a circularization reaction. The circular molecule is then processed using restriction enzymes or fragmentation. Fragments are enriched for the linker and outer library adapters are added around the two combined molecule ends. The internal adapter can then be used as a second priming site for an additional sequencing reaction in the same orientation or sequencing can be performed from the second adapter, from the reverse strand. (From “Understanding and improving high-throughput sequencing data production and analysis”, Ph.D. dissertation by Martin Kircher)
Thus in both cases (paired-end and mate-pair) a single physical piece of DNA (or RNA in the case of RNA-seq) is sequenced from two ends and so generates two reads. These can be represented as separate files (two FASTQ files with first and second reads) or a single file were reads for each end are interleaved (discussed later). Each of our data sets is a pair of FASTQ files, with each data set having a file for “Forward” read and “Reverse” reads.
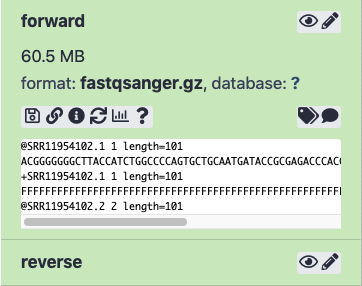
For example, the first two reads of the SRR11954102 data set are:
Forward
@SRR11954102.1 1 length=101
ACGGGGGGGCTTACCATCTGGCCCCAGTGCTGCAATGATACCGCGAGACCCACGCTCACCGGCTCCAGATTTATCAGCAATAAACCAGCCAGCCGGAAGGG
+SRR11954102.1 1 length=101
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF
@SRR11954102.2 2 length=101
GCATTAAGCGCGGCGGGTGTGGTGGTTACGCGCAGCGTGACCGCTACACTTGCCAGCGCCCTAGCGCCCGCTCCTTTCGCTGTCTTCCCTTCCTTTCTCGC
+SRR11954102.2 2 length=101
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFFFFFFFFFFFFFFFFFF
Reverse
@SRR11954102.1 1 length=101
AACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGTAGCAATGGCAACAACGTTGCGCAAAC
+SRR11954102.1 1 length=101
FFFFFFF:FFFFFFFFFFFFF:FFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFF
@SRR11954102.2 2 length=101
CCCTTTAGGGTTCCGATTTAGTGCTTTACGGGGAAAGCCGGCGAACGTGGCGAGAAAGGAAGGGAAGAAAGCGAAAGGAGCGGGCGCTAGGGCGCTGGCAA
+SRR11954102.2 2 length=101
FF:FFFFFFFFFFFFFFFFFF:FFFFFFFFF:FFFF:FFFFFFFFFFF:FFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFF
Key Points
Data can be found on NCBI using the a variety of identifiers for Bioproject, Biosample and SRA
Galaxy is an open source tool for conducting bioinformatic analyses
FASTQ files generally store raw sequencing data